-Enfermedad de Gaucher: presenta acumulación de sustancias grasosas en el bazo, hígado, pulmones, huesos y, a veces, en el cerebro.
Existen tres tipos: las opciones de tratamiento. Para los tipos 1 y 3 incluyen medicinas y terapia de sustitución de las hormonas y enzimáticas (imiglucerasa, velaglucerasa o taliglucerasa), que suele ser muy eficaz. No hay un buen tratamiento para el daño cerebral causado en los tipos 2 y 3.
-Enfermedad de mucopolisacaridosis: caracterizada por la ausencia o el mal funcionamiento de ciertas enzimas necesarias para el procesamiento de moléculas presentes en cada una de nuestras células. Estas ayudan a construir los huesos, cartílagos, tendones, córneas, la piel, el tejido conectivo y el tejido hematopoyético.
Existen varios tipos de mucopolisacaridosis; entre los más representativos están: la mucopolisacaridosis tipo I, también denominada gargolismo o enfermedad de Hurler. Estos niños pueden vivir hasta la adolescencia y presentan talla baja, deformidades óseas, retraso mental, hepatomegalia, alteraciones oculares y facies de gárgola. El tratamiento se realiza con terapia de sustitución enzimática con Laronidasa.
La enfermedad de Pompe: provoca una acumulación creciente de glucógeno en el lisosoma, que afecta, principalmente, al tejido muscular. En niños destaca por producir insuficiencia cardíaca al acumularse en el músculo cardíaco, causando cardiomegalia.
El tratamiento para las complicaciones cardíacas y respiratorias es tratarlas sintomáticamente. Terapias físicas y ocupacionales pueden ser beneficiosas para algunos pacientes. Las alteraciones en la dieta proporcionan mejoras temporales pero no alterarán el curso de la enfermedad. El asesoramiento genético provee a las familias la información con respecto al riesgo en los embarazos futuros.
La terapia de reemplazo enzimático se hace con alglucosidasa alfa.
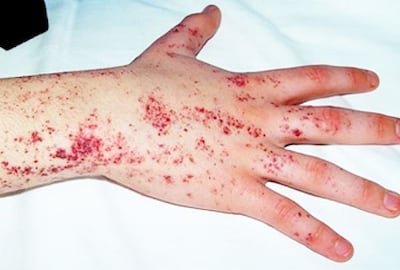
-La enfermedad de Fabry: en ella existe almacenamiento lisosómico hereditario, ligado al cromosoma X. Lamentablemente en la actualidad la enfermedad de Fabry no tiene cura, pero se ha conseguido un tratamiento de sustitución enzimática con agalsidasa alfa y agalsidasa beta.